A team of scientists from the United States and Sweden used state-of-the-art molecular analysis to explore the microbial environment on the International Space Station (ISS).

The International Space Station is featured in this image photographed by an STS-132 crew member on space shuttle Atlantis after the station and shuttle began their post-undocking relative separation. Undocking of the two spacecraft occurred on May 23, 2010, ending a seven-day stay that saw the addition of a new station module, replacement of batteries and resupply of the orbiting outpost. Image credit: NASA.
The results of their study, which is published in the journal Microbiome, provide strong evidence that specific human skin-associated bacteria make a substantial contribution to the space station microbiome (ecosystem of microorganisms in a particular environment).
“By using both traditional and state-of-the-art molecular analysis techniques we can build a clearer picture of the ISS’s microbial community, helping to spot bacterial agents that may damage equipment or threaten astronaut health, and identify areas in need of more stringent cleaning,” said Dr Kasthuri Venkateswaran, a senior research scientist at NASA’s Jet Propulsion Laboratory and a member of the Biotechnology and Planetary Protection Group.
Traditional microbiology techniques, which culture bacteria and fungi in the lab, have previously been used to assess the composition of the ISS microbiome.
Dr Venkateswaran and his colleagues have now used the latest DNA sequencing technologies to rapidly and precisely identify the microorganisms present on the station, filling in the gaps left by traditional methods.
The scientists collected air filter samples and vacuum bag dust from the ISS. They then compared these samples with dust from NASA cleanrooms, environmentally controlled and closed built spaces on Earth.
“Samples collected from the ISS and two cleanrooms at NASA’s Jet Propulsion Laboratory were analyzed by traditional cultivation, adenosine triphosphate, and propidium monoazide-quantitative polymerase chain reaction assays to estimate viable microbial populations. The 16S rRNA gene Illumina iTag sequencing was used to elucidate microbial diversity and explore differences between ISS and cleanroom microbiomes,” the scientists said.
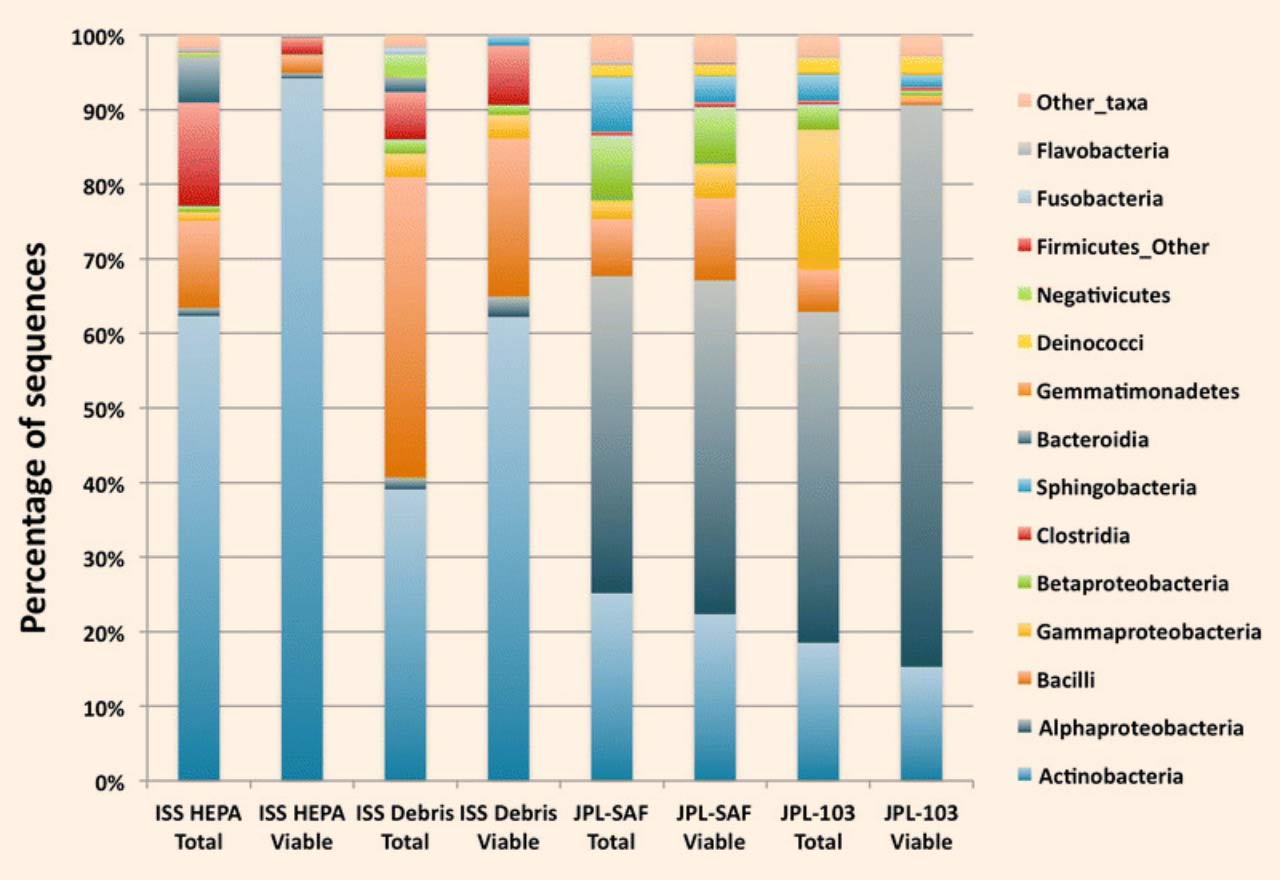
The samples analyzed during this study were the following: ISS HEPA filter particulates, vacuum cleaner bag components of ISS (ISS Debris), JPL Class 10 K cleanroom (JPL-SAF Debris), and JPL Class 1 K cleanroom (JPL-103 Debris). Among the ISS-associated samples, profiles were dominated by Actinobacteria, Bacilli, and Clostridia, while samples from the JPL-associated sites maintained higher levels of Alphaproteobacteria and Gammaproteobacteria. Image credit: Aleksandra Checinska et al.
“Statistical analyses showed that members of the phyla Actinobacteria, Firmicutes, and Proteobacteria were dominant in the samples examined but varied in abundance.”
“Actinobacteria were predominant in the ISS samples whereas Proteobacteria, least abundant in the ISS, dominated in the cleanroom samples.”
Key differences between the cleanrooms and the space station are the cleanrooms circulate fresh air while the ISS filters and recirculates air, and the ISS is inhabited continuously with only six people while 50 people may be in a cleanrooms in a day but not inhabit it continuously. Whilst these cleanrooms are not air-tight, they have several layers of rooms that would prevent free exchange of air particulates.
The findings help NASA establish a baseline for monitoring the cleanliness of the ISS, which will in turn help manage astronaut health in the future. However, since the study is based on genetic analysis, it could not conclude whether these bacteria are harmful to astronaut health.
_____
Aleksandra Checinska et al. 2015. Microbiomes of the dust particles collected from the International Space Station and Spacecraft Assembly Facilities. Microbiome 3: 50; doi: 10.1186/s40168-015-0116-3