
A team of scientists led by Dr. Massimo Ciccozzi from the University Campus Bio-Medico in Rome, Italy, has modeled the evolutionary development and diversity of Zika virus to understand how infection spreads between populations and how the virus reacts with the immune system.
Transmission electron micrograph of Zika virus, which is a member of the family Flaviviridae. Virus particles are 40 nm in diameter, with an outer envelope, and an inner dense core. Image credit: Cynthia Goldsmith / CDC.
Zika virus was accidentally discovered in Uganda in 1947, during a course of surveillance on mosquitoes and primates. It has now circled the globe, arriving in the Americas and in 2015, in the country of Cape Verde in West Africa.
This virus is the last of the long list of emerging flaviviruses that have recently received global attention due to the outbreak in Brazil and the rapid spread in several countries after the first detection in May 2015 in Brazil.
“Understanding the differences and similarities between Zika and other flaviviruses, such as the dengue fever and chikungunya viruses, is essential if effective drugs, vaccines and Zika-specific immunological tests for large population screening are to be designed,” Dr. Angeletti and co-authors said.
They carried out evolutionary analysis of Zika virus combined with homology (shared ancestry) modeling and T- and B-cells epitope prediction, which aims to determine how immune system responses cause the virus to react and change.
Their analysis revealed two distinct genotypes of the virus, African and Asiatic, and two separate clades.
“The phylogenetic results indicate two separate clades, the African one (clade I) containing the Uganda sequence as the most probable ancestor of Zika virus, and the clade II which contains all the most recent sequences isolated from the equatorial belt,” the authors explained.
“In this last clade, it is evident how in the recent past Cambodian strains diverged from the Malaysian ones. It is probable that the Cambodian strain was recently introduced or circulated in the past in this region but remained undetected until 2010, the date of isolation.”
“Interestingly, the Brazilian isolates clustered all together and appeared closely related to the French Polynesia isolates, indicating a probable relationship with strains causing outbreaks in other regions but in the same tropical belt.”
This lends support to the hypothesis that the virus might have been introduced to Brazil during the Va’a World Sprint Canoeing Championship in Rio de Janeiro in 2014, which included a team from French Polynesia, rather than the World Cup in which no teams from Pacific countries participated.
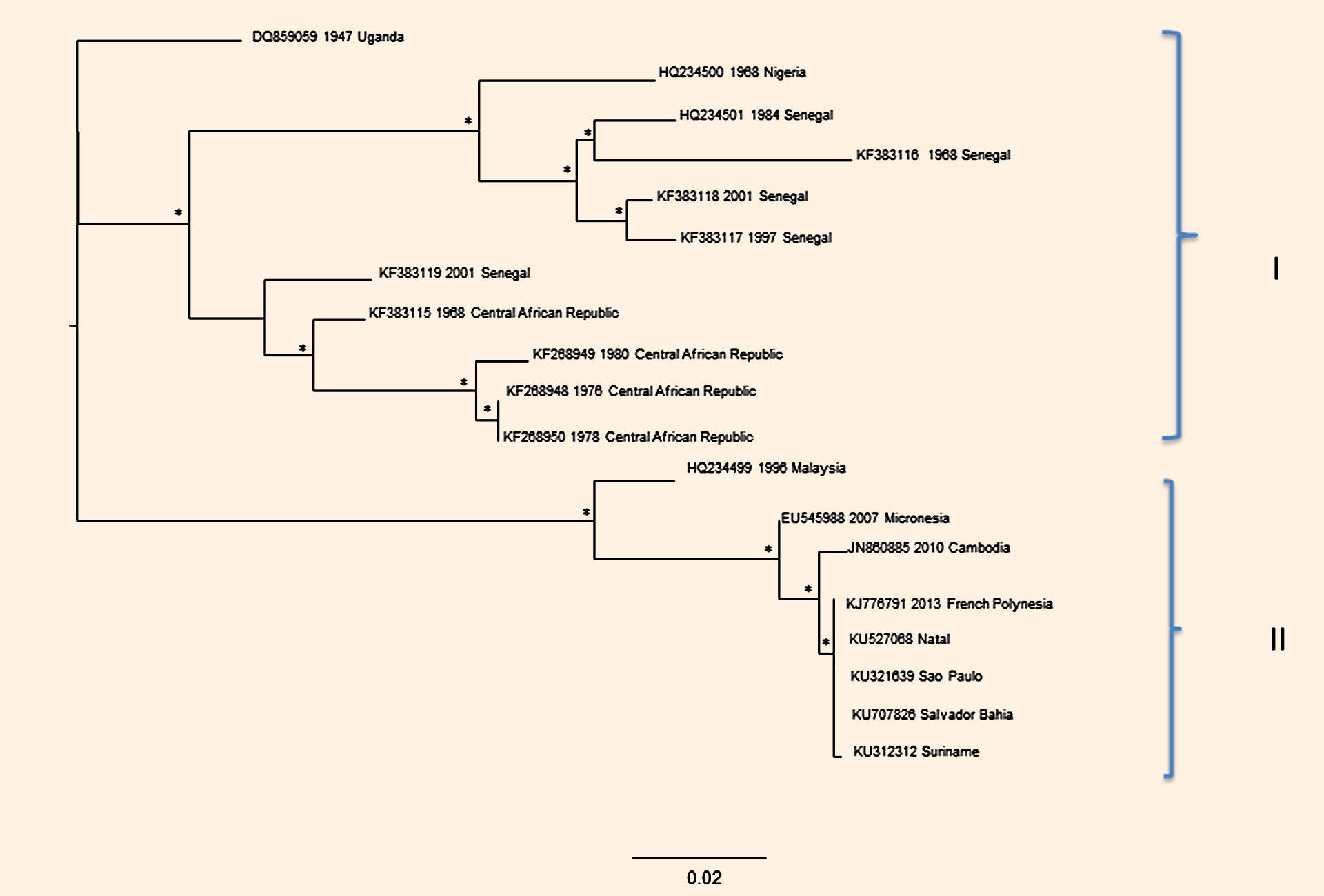
Phylogenetic trees of Env gene sequences of Zika virus. Image credit: Silvia Angeletti et al.
Among the factors that influence Zika infection, ‘antigenic variability’ — the way the virus alters its surface proteins to evade the host’s immune response — and pre-existing immunity caused by cross-reactions with other viruses might play an important role.
Such cross-reactions also make diagnosis of Zika infection unreliable, and could thus facilitate the spread of the virus.
The findings were published online in the journal Pathogens and Global Health on September 27, 2016.
_____
Silvia Angeletti et al. Phylogenesys and homology modeling in Zika virus epidemic: food for thought. Pathogens and Global Health, published online September 27, 2016; doi: 10.1080/20477724.2016.1235337






